

新藥查驗登記的核准,代表國家對此新藥品質、安全與療效的保證。在藥廠及藥商方面,若獲得上市的許可,藥品就可以在這個國家開始行銷販售,過往所投入的資金得以開花結果。醫藥品攸關人民身體健康及生命安全,受到各國政府的嚴格管制,因此新產品開發成功的關鍵常是在於能否通過政府衛生主管機關的上市審查。
在我國,藥物食品管理署 (TFDA) 是負責管理藥品、制訂政策的中央主管機關,同時也是檢驗藥品品質的執行機關。審查的基本原則是要證明此新產品是安全、有效、對病人是利多於弊的。食品藥物管理局 ( TFDA ) 有許多不同專業的人員參與,針對藥物的化學製造、藥理、藥動、毒理、臨床試驗都會有詳細的評審。
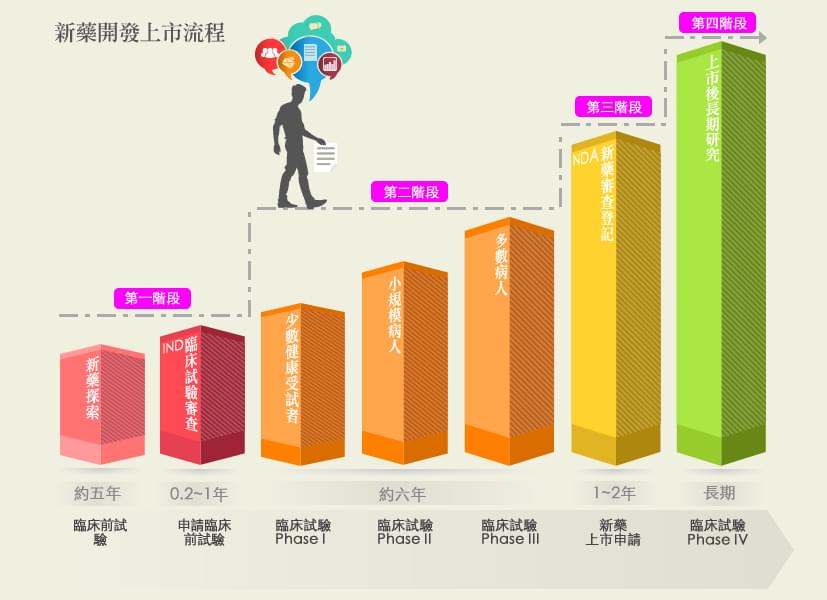
新藥的定義,依據藥事法第七條乃新成份、新療效複方或新使用途徑的藥品。本篇將簡述新藥上市許可的審核流程,主要可分為四階段:

當一個有療效的新成份開發出來並完成確效,蒐集所有研發之實驗資料向藥物管理局提出IND申請。需要送審的資料通常包括:藥物的來源組成、製造方法與規格、藥理與毒理之各種動物實驗、藥物動力學、臨床試驗計畫書與執行者等。上述藥理試驗及毒性試驗應符合優良實驗室規範(Good Laboratory Practice,GLP),可用來確保所製造或產品或所做之實驗的品質與再現性。
在台灣及美國,如果沒有太大問題,通常在 IND 建檔 (filing) 當天算起 30 天內,FDA 審查官認為合理的安全性沒有問題,即表示業者可以開始進行臨床試驗。

試驗中新藥必須通過臨床試驗前三期 (Phase I ~ Phase III),才會被允許提出新藥上市許可申請 (NDA)。如在第一、第二或第三期臨床試驗時發現新藥毒性太大,對人體造成重大傷害、新藥的規格及介紹錯誤不全、臨床試驗執行有誤、臨床試驗計劃有缺失…等,衛生主管機關可以視情況之嚴重性,要求暫時停止甚或中止臨床試驗。
臨床試驗的執行都應在衛生主管機關核可之醫學中心或醫院執行,而且必須經由人體試驗倫理委員會(IRBs)之審核及監督,以保障人體試驗的品質及受試者的安全與人權,確保參與者為志願參加,且研究人員確實有採取適當的措施保護受試者免於傷害,整個臨床試驗的過程皆符合優良臨床試驗規範(Good Clinical Practice, GCP)。

若是第三期臨床試驗結果顯示該新產品安全且有效,則廠商可準備所有相關資料:藥物對人體的作用及藥物製造程序之資料外,及所有動物、人體試驗資料和數據分析結果等等,向藥物食品管理署 (TFDA) 申請新藥上市許可。NDA 的審查,由於資料龐大,且需在藥效與安全之多方面考慮才准上市,因此審核相當費時。
在美國,當 NDA 申請案送到 FDA 時,FDA 有 60 天的時間決定是否對申請案進行建檔 (filing) 以便作進一步的審查;FDA 可以拒絕接受文件不完整或缺少某些試驗項目的申請案。對一般申請案,在接到案件後 10 個月內須完成 90%的審查,對於優先審查案則須於 6 個月內完成 90%的審查。

新藥上市後,就進入臨床試驗第四期(Phase IV):新藥監視期的評估與追蹤,以瞭解該新藥在廣大的人口使用下之安全性、有效性及可能之不同用途。如果發現有以前未發現的副作用,廠商亦必須向衛生主管機關報告,並加註在仿單內。嚴重時,衛生主管機關會撤銷該藥之上市許可。
另外,新藥上市後,衛生主管機關也會定期派員作實地查廠,繼續監督工廠操作是否符合優良製造規範。衛生主管機關對生物技術藥品及其他生物製劑有特別的規定:廠商須將每一批號產品送往主管機關抽樣檢查其是否符合品質規格,檢查通過後,才能上市販售。 一個新的藥品上市的過程,除了必須經過嚴格的重重試驗,也必須有完整的資料,依法審核之後才能上市,可謂花錢耗時又費力的產業。